methylTFR: Quantification of DNA Methylation Signatures in Transcription Factor Binding Sites
Irem B. Gündüz, Sarath Kumar Murugan, Fabian Muller
2026-07-22
Source:vignettes/methylTFR.Rmd
methylTFR.RmdIntroduction
DNA methylation can modulate transcription factor (TF) binding,
particularly when occurring within transcription factor binding sites
(TFBS). methylTFR is an R package that identifies DNA
methylation signatures at TFBS using whole-genome bisulfite sequencing
(WGBS) data.
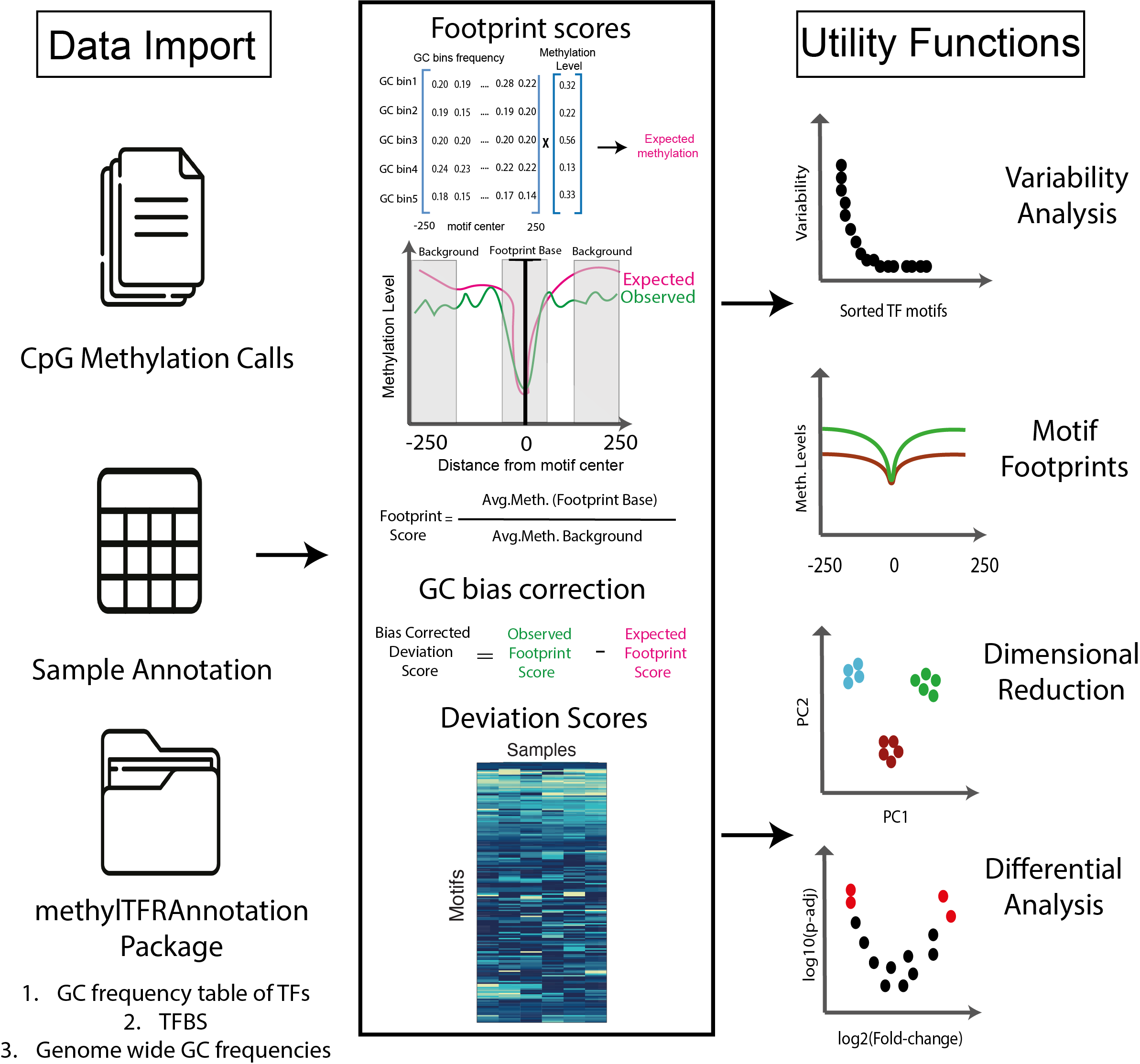
For each sample, methylation levels are first aggregated across all
genomic regions corresponding to TFBS. This yields the observed
deviation, which captures the raw signal of methylation
enrichment or depletion for each TF. To account for sequence composition
biases, methylTFR then estimates an expected
deviation using genomic background models derived from TF motif
GC content and genome-wide GC frequency. This deviation matrix provides
a compact and interpretable representation of TFBS methylation across
samples, suitable for downstream analyses such as dimensionality
reduction (e.g., PCA, UMAP), clustering, differential testing, and
visualization of TF-specific methylation footprints.
Installation
You can install the stable release of methylTFR from Bioconductor using:
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
BiocManager::install("methylTFR")To install the development version directly from GitHub:
if (!requireNamespace("remotes", quietly = TRUE)) {
install.packages("remotes")
}
remotes::install_github("EpigenomeInformatics/methylTFR")Getting Started
To get started with methylTFR, load the package and its dependencies:
library(methylTFR)
#> Loading required package: data.table
#> Warning: multiple methods tables found for 'sort'
#> Warning: multiple methods tables found for 'sort'
#> Warning: replacing previous import 'S4Arrays::read_block' by
#> 'DelayedArray::read_block' when loading 'HDF5Array'
#> Warning: replacing previous import 'S4Arrays::read_block' by
#> 'DelayedArray::read_block' when loading 'SummarizedExperiment'
#> Warning: multiple methods tables found for 'sort'
#>
#> Attaching package: 'methylTFR'
#> The following objects are masked from 'package:base':
#>
#> cbind, rbindRead a Sample File
The read_methylome() function is used to import
single-sample DNA methylation data into a GRanges
object.
It supports several common file formats, including EPP,
ALLC, BisSNP, bismarkCytosine,
bismarkcov, and ENCODE.
You can optionally filter out low-coverage sites using the
cov_threshold parameter (default = 1), which excludes
positions with insufficient read support.
Below is an example of reading an example EPP-formatted
file provided in the package:
epp_path <- system.file("extdata", "epp.tsv.gz", package = "methylTFR")
epp <- read_methylome(epp_path, "EPP")
epp
#> GRanges object with 6 ranges and 2 metadata columns:
#> seqnames ranges strand | score coverage
#> <Rle> <IRanges> <Rle> | <numeric> <numeric>
#> [1] chr1 3010957-3010958 + | 1.000 27
#> [2] chr1 3010959-3010960 - | 0.500 7
#> [3] chr1 3010971-3010972 + | 1.000 20
#> [4] chr1 3010973-3010974 - | 0.500 20
#> [5] chr1 3011025-3011026 + | 0.814 70
#> [6] chr1 3011027-3011028 - | 0.500 100
#> -------
#> seqinfo: 1 sequence from an unspecified genome; no seqlengthsAnnotation Resources
We provide precomputed TFBS-based annotation data for the human
genome (hg38), which is required by methylTFR to
compute expected deviations.
This includes transcription factor binding sites, GC content windows,
and motif GC frequency models.
You can download the hg38-compatible annotation package from the following AWS-hosted archive:
Optionally, you can create a customized annotation package for use
with methylTFR using the methylTFRAnnotationBuilder
package available on GitHub.
Input Data
methylTFR relies on several precomputed annotation
resources to estimate expected methylation levels at transcription
factor binding sites (TFBS). These annotations include:
-
GC distribution (
gcdist_subset): Genome-wide GC content distribution around cytosines, used to model methylation expectations. -
Motif GC frequency (
BATF_gcfreqs): GC frequency profile specific to the BATF motif across TFBS, used to correct for sequence composition bias. -
TF binding sites (
BATF_tf_bindsites): AGRangesobject containing the genomic coordinates of BATF binding sites. -
Example methylation data
(
example_data): A small subset of methylation calls in EPP format, read using theread_methylome()function, provided for demonstration purposes.
The following code loads these example datasets and displays the first few entries:
# Load the data
load(system.file("extdata", "gcdist_subset.rda", package = "methylTFR"))
load(system.file("extdata", "BATF_gcfreqs.rda", package = "methylTFR"))
load(system.file("extdata", "BATF_tf_bindsites.rda", package = "methylTFR"))
load(system.file("extdata", "example_data.rda", package = "methylTFR"))
# Check the data
head(gcdist)
#> GRanges object with 6 ranges and 2 metadata columns:
#> seqnames ranges strand | GC_bias GC_bin
#> <Rle> <IRanges> <Rle> | <numeric> <integer>
#> [1] chr1 10471-10500 * | 0.866667 5
#> [2] chr1 10591-10620 * | 0.633333 5
#> [3] chr1 10621-10650 * | 0.800000 5
#> [4] chr1 13051-13080 * | 0.700000 5
#> [5] chr1 13261-13290 * | 0.666667 5
#> [6] chr1 13291-13320 * | 0.600000 5
#> -------
#> seqinfo: 22 sequences from an unspecified genome
head(gcfreqs$BATF[, 1:5])
#> [,1] [,2] [,3] [,4] [,5]
#> [1,] 0.1398816 0.1394321 0.1386829 0.1366599 0.1361355
#> [2,] 0.1538173 0.1546415 0.1580880 0.1591369 0.1620589
#> [3,] 0.1962239 0.2001199 0.1965985 0.1973477 0.1916536
#> [4,] 0.2734697 0.2706226 0.2724957 0.2718214 0.2769911
#> [5,] 0.2366075 0.2351839 0.2341350 0.2350341 0.2331610
head(tf_bindsites)
#> $BATF
#> GRanges object with 268717 ranges and 1 metadata column:
#> seqnames ranges strand | score
#> <Rle> <IRanges> <Rle> | <numeric>
#> [1] chr1 47430-47840 + | 16.7687
#> [2] chr1 57232-57642 + | 13.2598
#> [3] chr1 93216-93626 + | 14.9499
#> [4] chr1 96525-96935 + | 13.6042
#> [5] chr1 99285-99695 + | 13.9006
#> ... ... ... ... . ...
#> [268713] chrY 57027771-57028181 - | 15.5241
#> [268714] chrY 57050236-57050646 - | 14.0836
#> [268715] chrY 57074721-57075131 - | 13.8672
#> [268716] chrY 57080582-57080992 - | 13.3138
#> [268717] chrY 57166552-57166962 - | 13.3138
#> -------
#> seqinfo: 24 sequences from an unspecified genome; no seqlengths
head(msites)
#> GRanges object with 6 ranges and 2 metadata columns:
#> seqnames ranges strand | score coverage
#> <Rle> <IRanges> <Rle> | <numeric> <integer>
#> [1] chr1 10471-10472 - | 1 9
#> [2] chr1 10608-10609 + | 0 2
#> [3] chr1 10609-10610 - | 1 1
#> [4] chr1 10616-10617 + | 0 2
#> [5] chr1 10617-10618 - | 1 1
#> [6] chr1 10619-10620 + | 0 2
#> -------
#> seqinfo: 170 sequences from an unspecified genome; no seqlengthsCompute Deviation Score for a Single Sample and Single Motif
The computeDeviation() function calculates the deviation
score for a specific transcription factor (TF) motif in a single sample.
It compares the observed methylation at TF binding sites (TFBS) to the
expected methylation derived from GC frequency models.
This example uses the BATF motif and the example
methylation dataset loaded earlier. The methylation data must first be
binned by GC content using addGCBintoMethylome() before
computing the deviation.
# Add GC bins to methylation data
bin_meth <- addGCBintoMethylome(msites, gcdist, ignoreStrand = TRUE)
bin_meth
#> gcbin avg_mscore
#> [1,] 1 0.5315789
#> [2,] 2 0.6466688
#> [3,] 3 0.7217031
#> [4,] 4 0.7091566
#> [5,] 5 0.7838198
# Compute deviation score for BATF motif
deviation_score <- computeDeviation(
motif = "BATF",
msites = msites,
tf_bindsites = tf_bindsites,
gcfreqs = gcfreqs,
enhancer = NULL,
ignoreStrand = TRUE,
binMsites = bin_meth
)
# View the result
deviation_score
#> dev exp_dev
#> <num> <num>
#> 1: 1.743674 0.9835985Run methylTFR on multiple samples and motifs
The methylTFR package provides a
run_methyltfr function to run the analysis on multiple
samples and motif sites. You need to download the annotation package for
the human genome (hg38) and place it in your working
directory.
library(methylTFRAnnotationHg38) # annotation package for hg38
gcfreqs <- getGCfreq(motifSet = "jaspar2020")
gc_dist <- getGenomeGC("hg38")
tf_bindsites <- getTFbindsites(motifSet = "jaspar2020")
sample_dir <- file.path("samples_dir")
sample_ann <- "samples.tsv" # should contain column name bedFile
# deviation score matrix
deviations <- run_methyltfr(sample_ann, # sample annotation file
sample_dir, # where the EPP files are
threads = 8, # number of threads
chunkSize = 10, # number of chunks to process
sampleColName = "bedFile", # column name for EPP file paths in sample_ann
tf_bindsites = tf_bindsites, # TF binding sites
gcfreqs = gcfreqs, # GC frequency
gc_dist = gc_dist, # GC distribution
filetype = "EPP" # file type
)Motif Footprinting
The methylTFR package provides a
plotMotifFootprint function to visualize the methylation
footprint of a specific TF motif per sample.
library(ggplot2)
load(system.file("extdata", "msites_sub.rda", package = "methylTFR"))
load(system.file("extdata", "gcdist_BATF.rda", package = "methylTFR"))
p <- plotMotifFootprint(
motif = "BATF",
tf_bindsites = tf_bindsites,
gc_dist = gcdist,
gcfreqs = gcfreqs,
msites = msites_sub,
sample_name = "ExampleSample",
enhancer = NULL,
method = "division"
)
p
#> Warning: Removed 112 rows containing missing values or values outside the scale range
#> (`geom_line()`).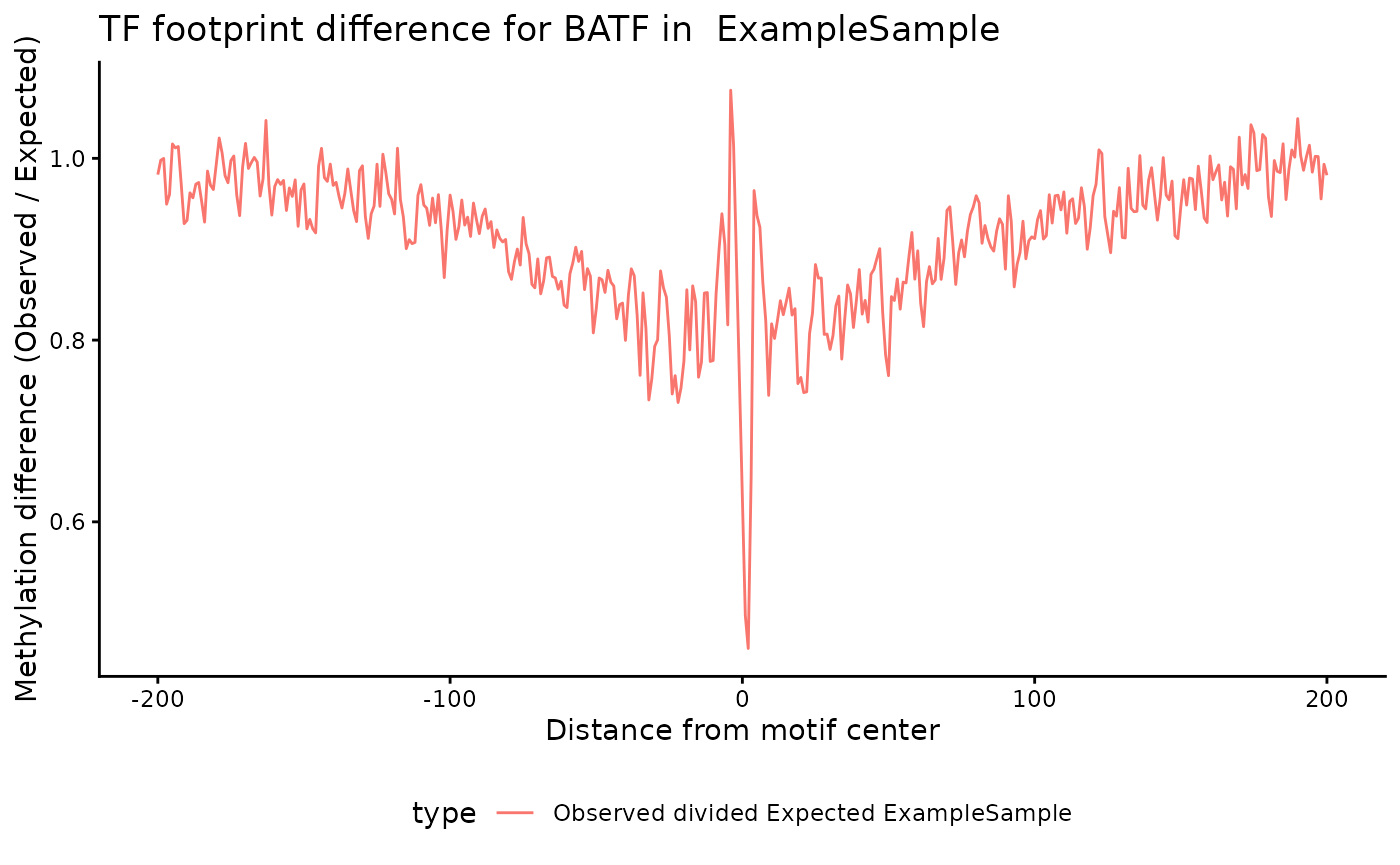
Expected vs Observed Footprint
To visually assess how observed methylation patterns compare with
expected profiles at transcription factor binding sites (TFBS),
methylTFR provides the plotExpectedFootprint()
function. This generates a footprint plot for a given motif in a single
sample, allowing you to inspect local methylation behavior relative to
GC-derived expectations.
In the example below, we visualize the methylation footprint for the
BATF motif using the included example data.
# Generate footprint plot for BATF
p <- plotExpectedFootprint(
motif = "BATF",
tf_bindsites = tf_bindsites,
msites = msites_sub,
sample_name = "ExampleSample",
gc_dist = gcdist,
gcfreqs = gcfreqs,
enhancer = NULL,
returnPlotData = FALSE
)
p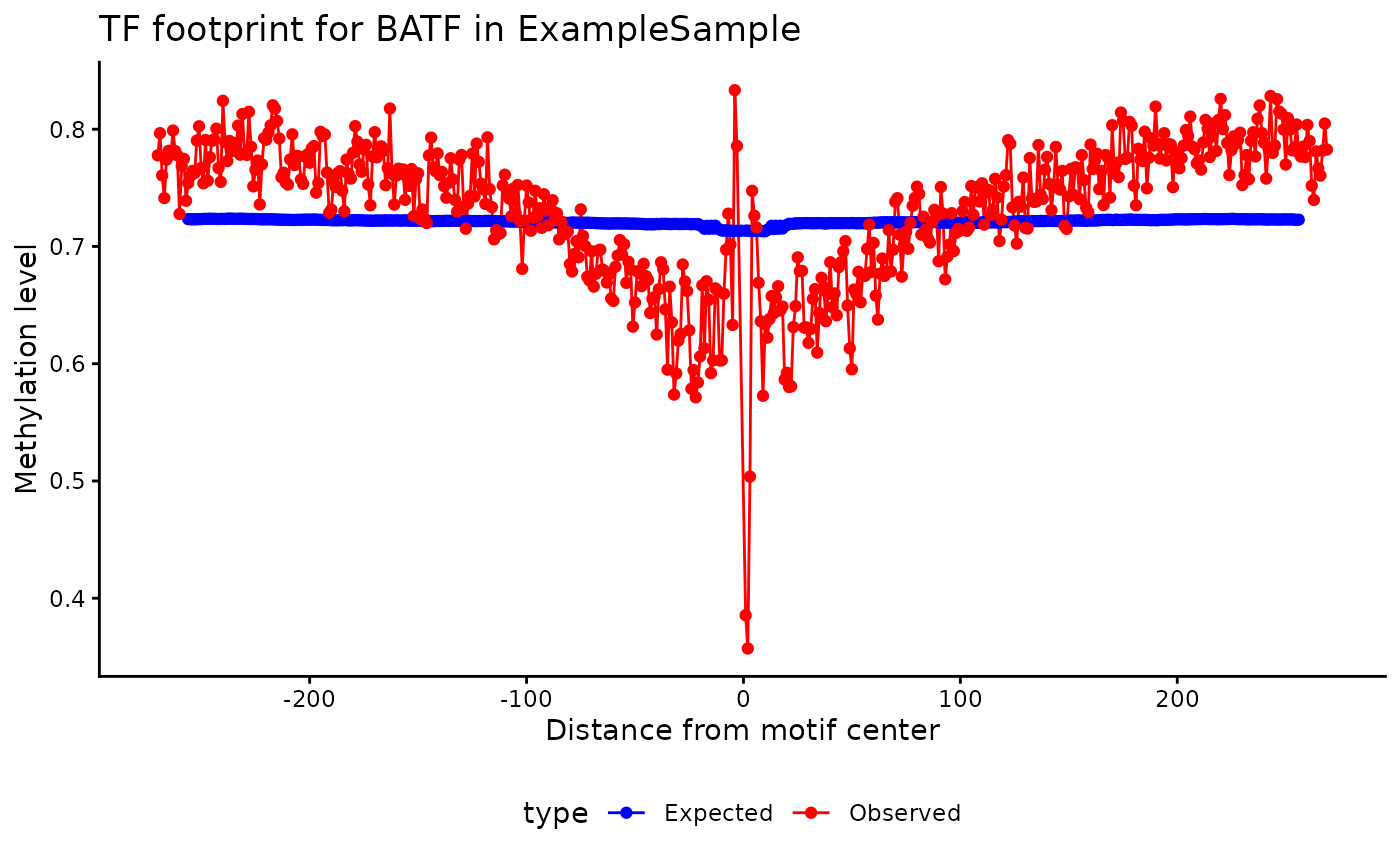
Differential TF Activity Analysis
To assess differential TF activity between two groups of samples,
methylTFR provides the
differential_deviation_test() function. This function
computes the observed and expected deviation scores for each TF motif
across the specified groups, enabling statistical testing to identify
differentially active TFs.
This is how methylTFRdeviations object is looked
like:
load(system.file("extdata", "tc_mem.rda", package = "methylTFR"))
load(system.file("extdata", "tc_naive.rda", package = "methylTFR"))
devs <- cbind(tc_mem, tc_naive)
devs
#> class: methylTFRdeviations
#> dim: 10 10
#> metadata(0):
#> assays(2): deviations z
#> rownames(10): FOXF2 FOXD1 ... RORA RORA(var.2)
#> rowData names(1): motifs
#> colnames(10): Tc-Mem_OP_S5_Long_D1.bedGraph.bed
#> Tc-Mem_OP_S4_Long_D60.bedGraph.bed ...
#> Tc-Naive_OP_S4_Long_D1.bedGraph.bed
#> Tc-Naive_OP_S3_High_D1.bedGraph.bed
#> colData names(4): CommonMinID condition cell_type bedFile
# Construct group labels from sample names
get_groupname <- function(x) {
return(unlist(strsplit(x, split = "_"))[1])
}
groups <- sub(
".bedGraph", "",
unlist(lapply(
FUN = get_groupname,
X = colnames(devs)
))
)
table(groups)
#> groups
#> Tc-Mem Tc-Naive
#> 5 5
# Run the differential deviation test
tc_result <- differential_deviation_test(
deviations = devs,
groups = groups,
alternative = "two.sided",
parametric = TRUE,
padjMethod = "BH"
)
# View the results
head(tc_result)
#> motifs p_value p_value_adjusted mean_difference
#> FOXF2 FOXF2 1.694736e-02 0.0355317162 0.02920759
#> FOXD1 FOXD1 3.644010e-01 0.4555013119 0.01081636
#> IRF2 IRF2 1.873667e-03 0.0093683362 0.03414276
#> MZF1(var.2) MZF1(var.2) 1.776586e-02 0.0355317162 0.01850831
#> MAX::MYC MAX::MYC 1.078111e-05 0.0001078111 0.09341429
#> PPARG PPARG 3.996324e-03 0.0133210805 0.03428684Session Information
sessionInfo()
#> R version 4.4.1 (2024-06-14)
#> Platform: x86_64-conda-linux-gnu
#> Running under: Debian GNU/Linux 11 (bullseye)
#>
#> Matrix products: default
#> BLAS/LAPACK: /icbb/projects/share/software/packages/miniconda3/envs/igunduz/lib/libopenblasp-r0.3.21.so; LAPACK version 3.9.0
#>
#> locale:
#> [1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
#> [3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
#> [5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
#> [7] LC_PAPER=en_US.UTF-8 LC_NAME=C
#> [9] LC_ADDRESS=C LC_TELEPHONE=C
#> [11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: Europe/Berlin
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.1 methylTFR_0.99.0 data.table_1.17.2 BiocStyle_2.34.0
#>
#> loaded via a namespace (and not attached):
#> [1] SummarizedExperiment_1.32.0 gtable_0.3.6
#> [3] xfun_0.52 bslib_0.9.0
#> [5] htmlwidgets_1.6.4 rhdf5_2.46.1
#> [7] Biobase_2.62.0 lattice_0.22-5
#> [9] generics_0.1.4 rhdf5filters_1.14.1
#> [11] vctrs_0.6.5 tools_4.4.1
#> [13] bitops_1.0-9 parallel_4.4.1
#> [15] stats4_4.4.1 tibble_3.2.1
#> [17] pkgconfig_2.0.3 R.oo_1.27.1
#> [19] Matrix_1.6-1.1 RColorBrewer_1.1-3
#> [21] S7_0.2.0 desc_1.4.3
#> [23] S4Vectors_0.40.1 lifecycle_1.0.4
#> [25] GenomeInfoDbData_1.2.13 stringr_1.5.1
#> [27] compiler_4.4.1 farver_2.1.2
#> [29] textshaping_0.4.0 GenomeInfoDb_1.42.3
#> [31] htmltools_0.5.8.1 sass_0.4.10
#> [33] RCurl_1.98-1.13 yaml_2.3.10
#> [35] pillar_1.10.2 pkgdown_2.2.0
#> [37] crayon_1.5.3 jquerylib_0.1.4
#> [39] R.utils_2.13.0 DelayedArray_0.28.0
#> [41] cachem_1.1.0 abind_1.4-8
#> [43] tidyselect_1.2.1 digest_0.6.37
#> [45] stringi_1.8.4 dplyr_1.1.3
#> [47] bookdown_0.43 labeling_0.4.3
#> [49] fastmap_1.2.0 grid_4.4.1
#> [51] cli_3.6.3 SparseArray_1.2.4
#> [53] magrittr_2.0.3 logger_0.4.0
#> [55] S4Arrays_1.6.0 dichromat_2.0-0.1
#> [57] withr_3.0.2 UCSC.utils_1.2.0
#> [59] scales_1.4.0 rmarkdown_2.29
#> [61] XVector_0.42.0 httr_1.4.7
#> [63] matrixStats_1.1.0 ragg_1.3.3
#> [65] R.methodsS3_1.8.2 HDF5Array_1.30.1
#> [67] evaluate_1.0.3 knitr_1.50
#> [69] GenomicRanges_1.54.1 IRanges_2.36.0
#> [71] rlang_1.1.4 glue_1.7.0
#> [73] BiocManager_1.30.25 BiocGenerics_0.52.0
#> [75] jsonlite_2.0.0 R6_2.6.1
#> [77] Rhdf5lib_1.28.0 MatrixGenerics_1.14.0
#> [79] systemfonts_1.2.3 fs_1.6.6
#> [81] zlibbioc_1.52.0